High-fidelity biomolecular modelling
An exciting seminar about combining evolutionary optimization algorithms with experimental, atomistic resolution target data to create a semi-automated tool for building high-fidelity molecular dynamics models.

Main content
Abstract: Although molecular dynamics (MD) simulations are a widely used tool in (bio)sciences, they often actually fail to provide a veritable description (e.g., matching nuclear magnetic resonnance (NMR) data within experimental accuracy) of the conformations and the dynamics of the molecules simulated—a crucial prerequisite for using MD to intuitively interpret experiments. These failures can largely be attributed to the quality of the underlying MD models (force fields), improvement of which is hindered by the lack of comprehensive comparison to experiments, and outdated approaches, such as hand-tuning parameters.
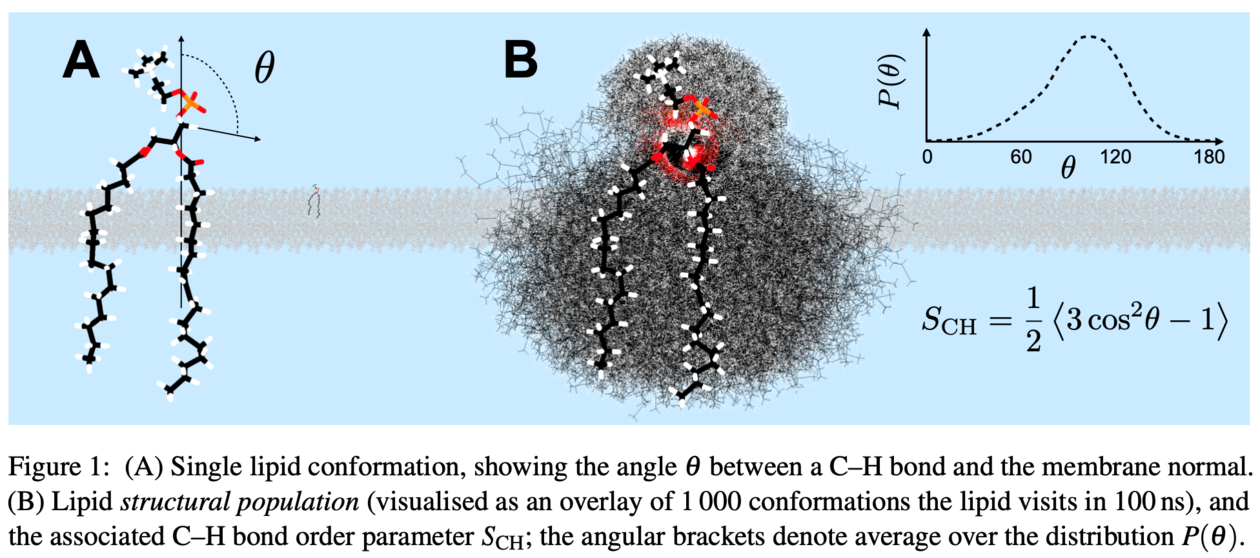
We address these issues by combining an evolutionary optimization algorithm with experimental, atomistic resolution target data (C–H bond order parameters measured with NMR) to create a semi-automated tool for building high-fidelity MD models. Using this approach, we have created the first phospholipid models that capture the conformational ensemble sampled by a bilayer lipid. They also represent well the lipid dynamics (as demonstrated by comparison to NMR relaxation times) and the overall bilayer structure (as quantified by comparison to small-angle x-ray scattering form factors).
As an application of this 'computational microscope', we quantify the conformational ensemble sampled by POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)—a demanding task for any other method outside simulations when considering an intrinsically disordered molecule—and compare the most prevalent conformations to the consensus structures predicted by the early pioneers of lipid NMR as well as the conformations present in crystalline state.
About the speaker: Markus Miettinen did his PhD in Theoretical and Computational Physics under the supervision of Mikko Karttunen at Aalto University, Helsinki, Finland. This was followed by post doctoral research in Germany in the groups of Zoya Ignatova (at Institute for Biology and Biochemistry, Potsdam University) and Roland Netz (at Department of Physics, Freie Universität Berlin), chiefly funded by the two independent post-doctoral grants he won from the European Molecular Biology Organization (EMBO Long-Term Fellowship 2011–2012) and the Volkswagen Foundation (VolkswagenStiftung Post-Doctoral Fellow 2012–2015). In 2016, he joined the Department of Theory and Bio-Systems lead by Reinhard Lipowsky at the Max Planck Institute of Colloids and Interfaces (Potsdam, Germany), where he was a research group leader until becoming an associate professor at UiB Chemistry and research group leader at CBU in 2022.