Mitochondria & Disease

Hovedinnhold
On mitochondria
Mitochondria are the energy producing elements of the cell. In the mitochondrial respiratory chain, basic nutrients like sugar and fat are converted to chemical energy in the form of adenosine triphosphate (ATP) by the process of oxidative phosphorylation (OXPHOS). In addition to energy production mitochondria are involved in numerous other processes such as protein catabolism detoxification (urea cycle), steroid and heme metabolism, calcium storage, apoptosis, etc
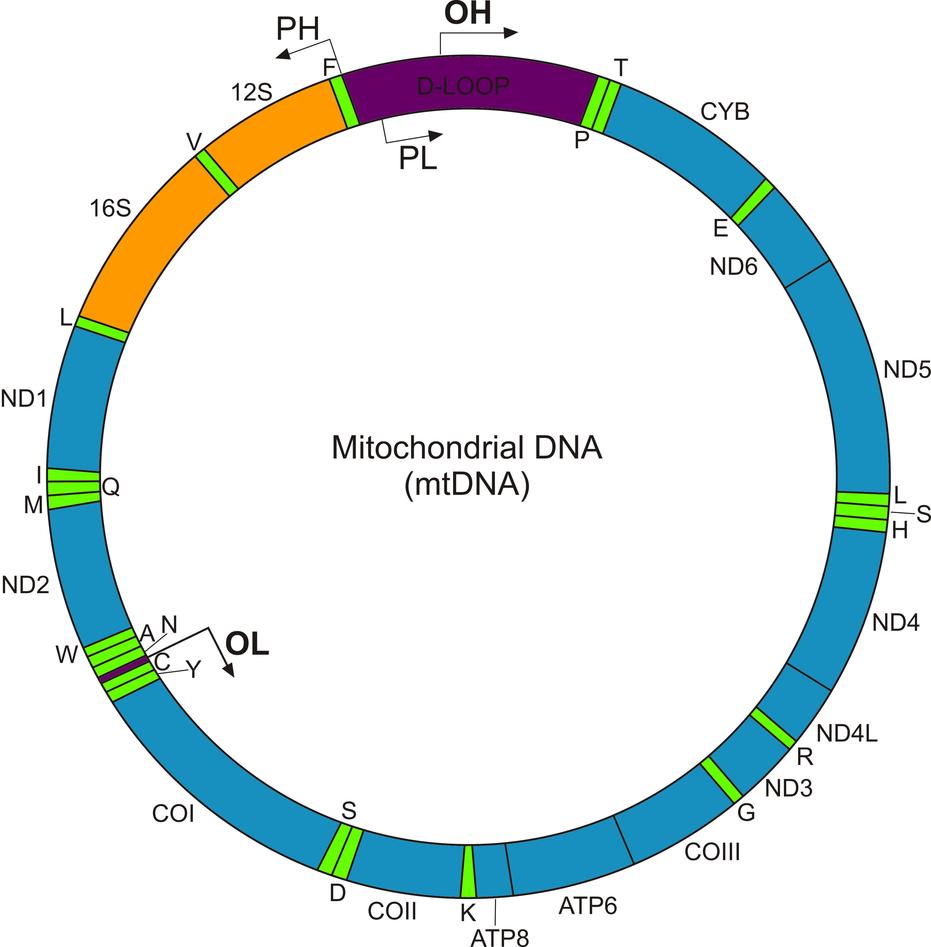
Mitochondria were once free living bacteria-like organisms and as a token of their past still carry their own genome, called mitochondrial DNA (mtDNA). MtDNA is a multi-copy genome found in up to thousands of copies per cell and is inherited exclusively from the mother. It comprises thirteen protein coding genes, which encode thirteen subunits of the respiratory complexes I, III, IV and V. The remaining (>80) respiratory chain subunits are encoded in the chromosomal genome together with thousands of other proteins involved in mitochondrial structure and function. Mitochondria (and chloroplasts in plants) are the only biological structures known to be under dual genetic control. Mitochondrial biogenesis and function depends on a continuous cross-talk between the nuclear and the mitochondrial genome.
Mitochondrial Disease
Mitochondrial disease may be broadly defined as any disorder caused by dysfunction of one of the many mitochondrial functions. In a medical context, however, the term mitochondrial disease is usually reserved for disorders affecting the respiratory chain. Such disorders may arise due to primary mutations of the mitochondrial DNA itself, or mutations in chromosomal genes that encode proteins involved in mitochondrial structure and function. In addition to directly causing disease, mitochondria also play an important, but still ill-defined role in most neurodegenerative disorders like motor neuron disease, dementia and parkinsonism.
Primary mitochondrial disease
Primary mitochondrial disease is caused by mutations of mtDNA. These include point mutations and large-scale rearrangements like deletions or duplications. Primary mtDNA mutations may occur sporadically or be inherited from the mother (maternal inheritance). A single case of paternal inheritance of a mitochondrial disorder has been reported.
Common examples of primary mitochondrial disease include chronic progressive external ophthalmoplegia (CPEO), Kearns-Sayre syndrome, mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes (MELAS), myoclonus epilepsy with ragged-red fibres (MERRF) and Leber’s hereditary optic neuropathy (LHON).
Nuclear mitochondrial disease
Nuclear mitochondrial diseases are caused by mutations in nuclear encoded proteins with mitochondrial localization and function. A few thousand proteins, most with still unknown function, are thought to localize in mitochondria. Members of the mitochondrial proteome are being increasingly recognised as the targets for disease causing mutations in humans. Some of the first to be recognised were proteins involved in the maintenance and replication of the mitochondrial genome like the DNA-polymerase gamma (POLG), which replicates and repairs mtDNA, or Twinkle, a mitochondrial helicase.
POLG mutations are a relativelly common cause of human genetic disease and are particularly abundant in Scandinavia. POLG mutations are associated with a broad spectrum of clinical disease ranging from late onset, mild myopathies to severe and often fatal infantile encephalopathy syndromes.